Co to jest zespół Marfana?
Zespół Marfana opisuje złożone zaburzenie dziedziczne tkanki łącznej, które dotyczy głównie oczu, układu sercowo-naczyniowego i układu mięśni szkieletowych. Jednak biorąc pod uwagę, że każdy narząd składa się z tkanki łącznej, zespół Marfana może idealnie zniszczyć i silnie zakłócić wzrost i funkcjonowanie każdego miejsca anatomicznego.
Zespół jest przenoszony jako cecha autosomalna dominująca: mamy zatem do czynienia z poważną chorobą genetyczną, mającą niezwykle zmienną ekspresję fenotypową (wady mogą się znacznie różnić od rodziny do rodziny lub od pacjenta do pacjenta).
To, co wywołuje zespół Marfana, to zmiana genu FBN1 (na chromosomie 15), który koduje fibrylinę-1, bardzo ważną glikoproteinę łączącą, która stanowi strukturalne wsparcie dla mikrofibryli.
Mikrofibryle: składające się z fibryliny, mikrofibryle są obecne w macierzy zewnątrzkomórkowej, w której tworzą wstęgę do osadzania elastyny we włóknach elastycznych. Chociaż wszechobecne w organizmie, mikrofibryle obfitują głównie w aortę, więzadła i zonety ciał rzęskowych (na poziomie oka).
Jako choroba autosomalna dominująca, tylko dzieci, które odziedziczyły gen FBN-1 zmieniony przez oboje rodziców, są dotknięte zespołem Marfana. Niemniej jednak w jednym przypadku spośród 4 choroba jest wynikiem spontanicznych mutacji u pacjentów, którzy nie mają historii rodzinnej.
Nazwa choroby pochodzi od francuskiego pediatry, który pierwszy opisał ją w 1896 r. (A. Marfan), po czym musiał czekać do 1991 r., Aby zidentyfikować zmieniony gen zaangażowany w objawową manifestację: odkrywcą był F. Ramirez.
Obejrzyj wideo
X Obejrzyj film na YouTubeprzyczyny
Wspomnieliśmy, że zespół Marfana jest bezpośrednią ekspresją mutacji genu kodującego fibrylinę-1. Spróbujmy pogłębić temat, aby zrozumieć, który mechanizm został ustanowiony, aby wywołać syndrom.
FIBRILLINA 1 to glikoproteinowy składnik elastyny, niezbędny do zapewnienia i utrzymania elastyczności i wytrzymałości tkanek. W warunkach fizjologicznych fibrylina 1 wiąże się z innym białkiem, znanym jako TGF-beta (lub transformujący czynnik beta wzrostu). Wydaje się, że TGF-beta bierze udział w szkodliwych procesach wpływających na mięśnie gładkie naczyń i macierz zewnątrzkomórkową. Wychodząc od tych założeń, niektórzy autorzy są przekonani, że syndrom Marfana jest spowodowany, oprócz mutacji genu FBN-1, także nadmiarem TGF-beta, zwłaszcza w aorcie, w zastawkach serca i płucach.
częstość
Szacuje się, że zespół Marfana dotyka 1 osobnika co 3000-5000 porodów i manifestuje się niewyraźnie między mężczyznami i kobietami, bez zamiłowania do precyzyjnej rasy. Statystyki pokazują, że 75% pacjentów ma pozytywną historię rodzinną; w pozostałych 25% przyczyną są sporadyczne mutacje, które wydają się być w jakiś sposób związane z zaawansowanym wiekiem ojca w momencie poczęcia.
Dzieci z wyjątkowo ciężkimi postaciami zespołu Marfana mają mniej niż rok życia.
Przed ewolucją strategii chirurgicznych na otwartym sercu większość pacjentów z zespołem Marfana miała średnią długość życia 32 lata; dzięki ciągłemu ulepszaniu terapii medycznych i farmakologicznych, obecnie pacjenci z zespołem Marfana żyją średnio do 60 lat.
Objawy i objawy
Aby dowiedzieć się więcej: Objawy Zespół Marfana
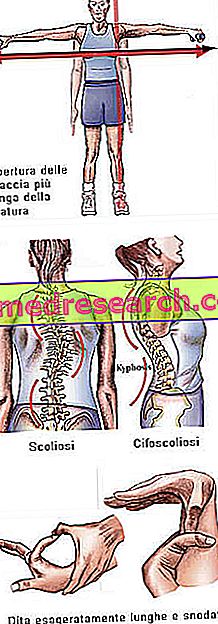
Zespół Marfana może przebiegać całkowicie bezobjawowo. Dotknięci chorobą pacjenci mają nadmiernie smukłą strukturę, nieproporcjonalnie wysoką i cienką. Kończyny dolne i górne mają znacznie większą długość niż pień (dolicostenomegalia). Mówi się również o arachnodactyly, aby najlepiej wyrazić koncepcję przesadnej długości palców, typowej dla osób dotkniętych syndromem Marfana: ręce są zatem porównywane do nóg pająka.
Jeśli chodzi o wzrost, pacjenci ci mają średni wzrost powyżej 97. percentyla.
Inne wyróżniające cechy często występujące u pacjentów z zespołem Marfana to:
- Ramiona otwarte powyżej wysokości
- Luźne połączenia → przesadna ruchomość stawów
- Deformacja ściany klatki piersiowej
- Przemieszczenie krystaliczne
- Górna część ciała mniej rozwinięta niż dolna część
- Spontaniczna odma opłucnowa (11%)
- skolioza
- Rozstępy skóry na udach, plecach, naramiennikach, poziomie piersiowym
Wśród najbardziej problematycznych objawów związanych z zespołem Marfana wspominamy wypadanie zastawki serca i niewydolność zastawki mitralnej: podobny stan może łatwo promować rozszerzenie pierścienia aorty i rozwarstwienie aorty.
Tabela pokazuje objawy, które można znaleźć u pacjentów z zespołem Marfana. Opisane tam postacie nie zawsze są obecne, ale można znaleźć znaczną ich część.
| Dotknięte miejsce anatomiczne | Możliwe objawy |
ładny | Rozstępy w okolicy piersiowej, lędźwiowej i krzyżowej |
oczy | Upośledzenie wzroku, astygmatyzm, odwarstwienie siatkówki, jaskra z wąskim kątem przesączania, zwichnięcie soczewki, krótkowzroczność |
Struktura kości | Bóle stawów, kifoskolioza, dolicostenomelia (nadmierny rozwój długości kończyny w porównaniu do tułowia), nadmierna ruchliwość, wysokie podniebienie, zdeformowana klatka piersiowa, płaskie stopy, wąskie i cienkie nadgarstki, nieprawidłowe ponowne wejście / wysunięcie mostka, skolioza, zakrzywione ramiona, kręgozmyk |
Dita | arachnodaktylia |
płuca | Spontaniczna odma opłucnowa, duszność, idiopatyczna obturacyjna choroba płuc |
Zmiany twarzy | Podniebienie ogival (wada podniebienia), retrognacja żuchwy (wada w rozwoju szczęki), wydłużona twarz |
serce | Dławica piersiowa, tętniak aorty brzusznej, zaburzenia rytmu serca, rozszerzenie aorty piersiowej / pęknięcie / rozwarstwienie, niewydolność aorty, wypadanie zastawki mitralnej |
język | Trudność językowa |
diagnoza
Biorąc pod uwagę ponad 200 możliwych mutacji, użycie markerów genetycznych jest prawie niemożliwe do celów diagnostycznych.
Ustalenie zespołu Marfana nie zawsze jest tak natychmiastowe, biorąc pod uwagę, że fenotypowa ekspresja mutacji nie zawsze jest oczywista i łatwa do zidentyfikowania. Opóźnienie diagnostyczne może poważnie zagrozić przeżyciu pacjenta: pomyśl na przykład o braku rozpoznania problemu sercowo-naczyniowego.
Kryteria diagnostyczne zespołu Marfana zostały opracowane na arenie międzynarodowej w 1996 roku: diagnoza polega na badaniu historii rodziny związanej z kombinacją głównych i niewielkich wskaźników zespołu.
Niektóre z wielu stosowanych testów diagnostycznych to:
- echokardiograficznego
- Angiorisonance magnetyczna i CT (do badania aorty)
- angiografia rezonansu magnetycznego (MRA) z płynem kontrastowym (w celu uwidocznienia wewnętrznych struktur aorty)
- badanie lampami szczelinowymi (w celu przeanalizowania możliwego przemieszczenia soczewki)
- pomiar ciśnienia w gałce ocznej (aby podkreślić możliwą obecność jaskry)
- testy genetyczne (zalecane przed poczęciem dziecka w celu ustalenia zespołu lub nie)
terapie
Jako choroba genetyczna nie ma leku ani leczenia, które mogłyby odwrócić chorobę.
Stosowanie leków jest jednak niezbędne, aby złagodzić objawy i uniknąć powikłań, zwłaszcza sercowych. W tym celu szczególnie odpowiednie są leki obniżające ciśnienie krwi, takie jak sartany (zwłaszcza), inhibitory ACE i beta-blokery.
W kontekście zespołu Marfana pacjenci cierpiący również na skoliozę mogą podchodzić do szczególnej opieki, jak również do osób dotkniętych jaskrą.
Operacja jest możliwa do skorygowania nieprawidłowego rozszerzenia aorty, elementu, który często łączy większość pacjentów z zespołem Marfana.
Kontynuuj: Zespół Marfana - narkotyki i opieka »


