Zespół Klinefeltera jest chorobą genetyczną, która dotyka tylko mężczyzn. Cechą charakterystyczną tej choroby jest obecność dodatkowego chromosomu X. Ten chromosom nie pozwala na normalny rozwój czysto męskich cech płciowych w okresie dojrzewania.
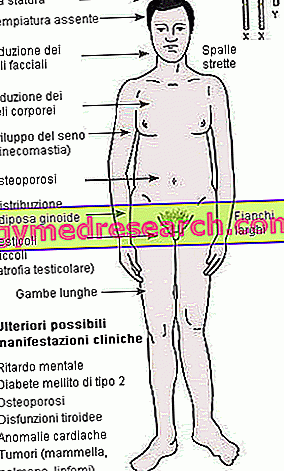
Rysunek: pokazuje podsumowanie głównych objawów klinicznych zespołu Klinefeltera. Od hipogonadyzmu do typowo kobiecych cech fizycznych (szerokie biodra, ginekomastia i wąskie ramiona), od upośledzenia umysłowego do cukrzycy.
Niestety, nie ma konkretnego lekarstwa. Jednak niektóre zabiegi terapeutyczne zmniejszają najcięższe objawy iw konsekwencji poprawiają jakość życia pacjentów.Ludzkie chromosomy
Każda komórka zdrowego człowieka zawiera 23 pary chromosomów. Para tych chromosomów ma charakter seksualny, to znaczy określa płeć jednostki; pozostałe 22 pary składają się z chromosomów autosomalnych. W związku z tym ludzki genom ma 46 chromosomów.
ZMIANY GENÓW
Każda para chromosomów zawiera pewne geny .
Gdy mutacja występuje w chromosomie, gen może być wadliwy. Ten wadliwy gen w konsekwencji wyraża uszkodzone białko.
Kiedy natomiast liczba chromosomów się zmienia, mówimy o aneuploidii . W tym przypadku zamiast dwóch chromosomy mogą być trzy (trisomia) lub tylko jeden (monosomy).
CHROMOSOMY SEKSUALNE
Chromosomy seksualne są niezbędne do określenia płci, mężczyzny lub kobiety. Kobieta ma w komórkach swojego ciała dwie kopie tak zwanego chromosomu X ; człowiek natomiast ma chromosom X i chromosom Y. Jeśli chodzi o chromosomy autosomalne, również te seksualne są dziedziczone po rodzicach: kopia jest podana przez ojca, inna przez matkę.
Istnieje kilka patologii genetycznych związanych z chromosomami płciowymi. Przedstawiają one zmiany w strukturze chromosomów lub zmiany w liczbie chromosomów. Wśród tych ostatnich, oprócz Klinefeltera, wspominamy o zespole turnera, zespole trisomii X i zespole podwójnego Y.
Czym jest zespół Klinefeltera
Zespół Klinefeltera jest chorobą genetyczną płci męskiej, charakteryzującą się obecnością chromosomu trzeciej płci (trisomii). Ten chromosom jest typu X. Komórki pacjentów z zespołem Klinefeltera mają zatem zestaw chromosomalny 47, XXY.
Mężczyźni z dodatkowym chromosomem X mają:
- Anomalie jąder (męskie gonady), zwane także dysgenezją jąder .
- Mało rozwinięte drugorzędne postacie seksualne.
- Kobiece postacie somatyczne.
epidemiologia
Zespół Klinefeltera dotyczy jednego mężczyzny na każde 500 nowych porodów. Dane są jednak niepewne. W rzeczywistości inne szacunki mówią o jednym przypadku na 1000 nowych urodzeń. Ta niepewność wynika z faktu, że niektóre łagodne formy choroby pozostają niezauważone, ponieważ określają prawie niezauważalne objawy.
Ponadto, co 100 nie płodnych mężczyzn, 3 mają zespół Klinefeltera.
przyczyny
Nieprawidłowa obecność chromosomu płciowego dodatkowego typu X determinuje zespół Klinefeltera u mężczyzn. Należy zauważyć, że jest to nie-dziedziczna anomalia genetyczna. Dlatego rodzice są zdrowi.
Dlaczego więc niektórzy synowie rodzą się z dodatkowym chromosomem X?
patogeneza
Odpowiedź z genetyczno-biologicznego punktu widzenia jest bardzo złożona. Wydaje się, że błąd genetyczny zaczyna się podczas mejozy lub podczas mitozy zapłodnionego jaja, czyli zarodka.
Mejoza to proces, w którym niektóre komórki ciała z 46 chromosomami dzielą się na 4 komórki płciowe z 23 chromosomami każda. Komórki te to plemnik dla człowieka i komórka jajowa ( oocyt ) dla kobiety. Połączenie komórki jajowej z komórką nasienia powoduje zapłodnienie komórki jajowej z 46 chromosomami (23 + 23).

Rysunek: pokazuje proces niezwiązania podczas mejozy. Jedna z czterech powstających komórek niesie ze sobą oba chromosomy. Jeśli ta komórka jest tą, która zapładnia jajo (lub jest to samo jajo), wtedy pojawi się anomalia genetyczna.
Mitoza to podział komórki macierzystej na dwie identyczne komórki potomne z kompletnym zestawem chromosomów.
Błąd genetyczny polega na braku rozdzielenia (lub braku dysjunkcji ) jednej z chromatyd, tj. Chromosomów w zduplikowanej formie. Dlatego, gdy komórki dzielą się:
- W mejozie komórka płciowa będzie miała liczbę chromosomów 24 chromosomów.
- W mitozie komórka potomna będzie miała chromosomową liczbę 47 chromosomów.
W przypadku zespołu Klinefeltera ten dodatkowy chromosom jest właśnie chromosomem X.
GENETYKA: XY / XXY MOSAICISM
Niektórzy pacjenci z zespołem Klinefeltera mają genom całkowicie 47, XXY. Z drugiej strony inni pacjenci mają mieszany genom: 47, XXY i 46, XY. W tym przypadku mówimy o mozaikowatości genetycznej .
Jakie jest to wszystko wyjaśnienie?
Wszystko zależy od momentu, w którym nastąpi niezwiązanie .
Jeśli występuje podczas mejozy, genom jest całkowicie 47, XXY. W rzeczywistości komórka jajowa lub plemnik przedstawia natychmiast zmienioną liczbę chromosomów.
Z drugiej strony genom jest mieszany, gdy niezwiązanie występuje podczas mitozy zapłodnionego jaja. W tym przypadku plemnik i komórka jajowa w momencie zapłodnienia posiadają normalną liczbę chromosomów. Błąd braku separacji występuje podczas podziału komórek zarodka. Wynik: pod koniec rozwoju embrionalnego niektóre komórki będą prezentować 46 chromosomów, inne 47.
Pacjenci z genomem XY / XXY zwykle mają osłabione objawy w porównaniu z tymi z genomem całkowicie 47, XXY.
GENETYKA: INNE WARIANTY
Chociaż rzadko, pacjentów obserwowano z zestawem chromosomalnym złożonym z więcej niż 2 chromosomów X. Na przykład:
- 48, XXXY
- 49, XXXXY
- 48, XXYY
- 49, XXXYY
Obecność tych genomów jest bardzo rzadka i często zbiega się z poważnymi formami deficytu intelektualnego i wieloma wadami fizycznymi.
objawy
Aby dowiedzieć się więcej: Objawy zespołu Klinefeltera
Pierwsze objawy zespołu Klinefeltera pojawiają się w okresie dojrzewania, wpływając na normalną ewolucję. W rzeczywistości proces dojrzewania cierpi na spowolnienie i wczesne zakończenie . Głównymi objawami klinicznymi są hipogonadyzm (tj. Małe jądra) i zmieniony proces dojrzewania plemników ( spermatogeneza ).
W wieku przedpokwitaniowym (niemowlęctwo i przed dojrzewaniem) tylko niektórzy pacjenci wykazują oznaki prowadzące do podejrzenia zespołu Klinefeltera. Są to raczej niejasne przejawy, ponieważ są one wspólne dla innych okoliczności patologicznych lub dlatego, że nie są tak oczywiste, aby przyciągnąć uwagę członków rodziny.
MOŻLIWE SYGNAŁY WIEKU? przedpokwitaniowy
Deficyty napotkane w tej fazie wzrostu mogą być następujące:
| deficyt | opis |
| Niewielkie trudności w nauce i problemy z uwagą | Upośledzenie umysłowe |
| Zmniejszona siła mięśni | Mięśnie są słabsze niż rówieśnicy |
| Dyspassia słowna i ruchowa | Trudność mowy i mowa Opóźnienie w podejmowaniu pierwszych kroków Brak koordynacji |
| dysleksja | Trudności z czytaniem |
| Problemy behawioralne | Pacjenci są zazwyczaj zamknięci w sobie i niepewni siebie. Wyglądają na niedojrzałe w porównaniu z rówieśnikami |
PO PUBLICIE?
W tym okresie życia symptomatologia zespołu Klinefeltera staje się oczywista. Główne objawy kliniczne to:
| Objaw / znak | opis | częstotliwość |
| Małe jądra | Jest to tak zwany hipogonadyzm | > 90% |
| bezpłodność | Ze względu na:
| > 90% |
| Zwiększona liczba gonadotropin we krwi i moczu | Gonadotropiny nie stymulują, jak powinny:
| > 80% |
| Redukcja testosteronu we krwi | Konsekwencja braku działania gonadotropin na komórki jąder, które wytwarzają testosteron. | > 80% |
| Zmniejszenie wzrostu włosów | Na poziomie:
| 80% 30% > 45% |
| ginekomastia | To dwustronny rozwój piersi | 50% |
| Problemy z erekcją | Towarzyszy im brak libido | > 60% |
| Zmniejszenie rozwoju penisa | > 20% | |
| Deficyt intelektualny | 10-20% |
Co więcej, nawet aspekt fizyczny zakłada specyficzne cechy:
- otyłość
- Wysoka postawa
- Kończyny górne i dolne są wydłużone i nie są proporcjonalne do reszty ciała
- Fizjonomia ciała podobna do kobiety (wąskie ramiona i szerokie biodra)
PROBLEMY PSYCHOLOGICZNE
Aspekt psychologiczny zasługuje na osobny rozdział. Pacjenci z zespołem Klinefeltera cierpią na depresję . W rzeczywistości dostrzegają różnice od zdrowych rówieśników iw konsekwencji reagują postawami charakteryzującymi się:
- introwercja
- uległość
- niepokój
Hipogonadyzm, brak libido i ginekomastia to główne czynniki wpływające na psychikę pacjentów z zespołem Klinefeltera.
POWIKŁANIA
Są one spowodowane głównie zaburzeniami hormonalnymi, które wpływają na testosteron. W rzeczywistości brak syntezy testosteronu zwiększa poziom cholesterolu krążącego we krwi i sprzyja osteoporozie . Co więcej, mężczyźni z zespołem Klinefeltera są bardziej dotknięci niż mężczyźni z rakiem piersi .
Wreszcie związek z cukrzycą jest bardzo powszechny.
| powikłanie | pogłębienie |
| Choroby układu krążenia | Z powodu hipercholesterolemii Leczenie testosteronem obniża poziom cholesterolu |
| osteoporoza | Ze względu na niski poziom testosteronu |
| Rak piersi | Z powodu ginekomastii. Ryzyko jest znacznie wyższe niż u zdrowych mężczyzn |
| Cukrzyca | Bardzo często |
| Skrzeplina-zator | Ze względu na tworzenie się skrzepów krwi w naczyniach krwionośnych |
| Choroby autoimmunologiczne |
|
diagnoza
Aby prześledzić obecność dodatkowego chromosomu X, stosuje się test genetyczny znany jako kariotyp .
kariotyp

Rysunek: zestaw chromosomów pacjenta z zespołem Klinefeltera obserwowany w teście kariotypu.
Polega na analizie zestawu chromosomów danej osoby.
Kariotyp ujawnia, czy występują zmiany w normalnej liczbie chromosomów. Można go wykonać na próbce płynu owodniowego, w prenatalnej diagnozie choroby lub na próbce krwi, w celu rozpoznania poporodowego.
Jak wspomniano powyżej, dojrzewanie jest kluczowym czasem dla wystąpienia objawów zespołu Klinefeltera. W poprzednich fazach (przed porodem i przed okresem dojrzewania) kariotyp jest jedynym testem diagnostycznym przydatnym do wykrywania zespołu Klinefeltera. Kiedy jest wykonywana, bardzo często z obawy przed innymi patologiami.
Po okresie dojrzewania kariotyp potwierdza diagnozę wstępną na podstawie objawów.
INNE PRZYDATNE BADANIA
Obok kariotypu istnieją inne bardzo przydatne i wskazujące badania choroby. Składają się z badań krwi i moczu, biopsji jąder i densytometrii kości.
Poniżej znajduje się tabela pokazująca test, dlaczego ma miejsce i inwazyjność.
| Test diagnostyczny | Dlaczego? | MICROdentistry |
| Badanie krwi | Aby ocenić poziomy:
| nie |
| Badanie moczu | Aby ocenić poziomy:
| nie |
| Biopsja jąder | Aby ocenić:
| Tak, minimalnie inwazyjny, to interwencja chirurgiczna; wymaga znieczulenia miejscowego; nie wymaga hospitalizacji pooperacyjnej. |
| Gęstość mineralna kości | Aby ocenić:
| nie |
Wreszcie, niektóre oceny aspektu fizyczno-anatomicznego, takie jak:
- Mały rozmiar jąder (hipogonadyzm).
- Niewielki porost włosów na twarzy, pachach i łonie.
terapia
Będąc chorobą genetyczną, nie ma lekarstwa, które rozwiązałoby główny problem. Można jednak zastosować pewne środki terapeutyczne, przydatne do:
- Prawidłowy i ograniczający hipogonadyzm
- Zmniejsz efekty ginekomastii
- Wspieranie płodności
TERAPIA HORMONALNA
Głównym lekarstwem dla pacjentów z zespołem Klinefeltera jest leczenie hormonalne oparte na testosteronie . Celem jest podniesienie niskiego poziomu we krwi.
Terapia hormonem testosteronu rozpoczyna się w okresie dojrzewania, aw niektórych przypadkach trwa całe życie.
Tabela ilustruje główne cechy leczenia hormonalnego.
| Charakterystyka leczenia testosteronem | opis |
| Po co to administrować? | Aby zwiększyć niski poziom testosteronu u pacjentów |
| Wpływ testosteronu |
|
| dawkowanie | 250 mg co 3-4 tygodnie |
| Brak efektu lub minimalny efekt |
|
LECZENIE GYNECOMASTII
Nieprawidłowy rozwój piersi często powoduje u pacjentów depresję i poczucie zakłopotania . Dlatego istnieje możliwość poddania się operacji w celu zmniejszenia objętości piersi. W rzeczywistości, tkanka tłuszczowa i gruczołowa jest eliminowana. To dość inwazyjna operacja.
LECZENIE STERYLNOŚCI? (Niepłodność?)
Pacjent z zespołem Klinefeltera, który chce mieć dzieci, powinien skontaktować się z genetykiem i specjalistą od niepłodności męskiej.
Pierwszy z nich proszony jest o ocenę, czy istnieje możliwość dziedzicznej transmisji choroby. Drugi ma za zadanie zbadanie zdolności spermatogenezy pacjenta, aby zrozumieć, czy istnieje konkretna możliwość poczęcia.
Jeśli nawet minimalna ilość plemników jest dojrzała i zdolna do zapłodnienia jaja (lub jajka), możemy przystąpić do:
- Zapłodnienie in vitro (IVF)
- Docytoplazmatyczna iniekcja plemnika (ICSI) w oocycie
LECZENIE PSYCHOLOGICZNE
Aspekt psychologiczny jest bardzo ważny. Podawanie testosteronu, chirurgiczna redukcja piersi i ewentualna płodność poprawiają poczucie własnej wartości u pacjentów z zespołem Klinefeltera. W rzeczywistości udaje im się pokonać poczucie wykluczenia z aktywnego życia społecznego i związku, które jest wspólne dla ich rówieśników.
INNE ZABIEGI
Niektórzy pacjenci doświadczają objawów przed okresem dojrzewania. Tabela pokazuje możliwe środki zaradcze, które należy podjąć w obecności niektórych z tych zaburzeń.
| Środki zaradcze i powiązane zakłócenia |
| Terapia językowa, aby poprawić trudności w wyrażaniu mowy |
| Fizjoterapia, aby poprawić brak koordynacji |
| Specjalistyczne leczenie dysleksji |
rokowanie
Opieka hormonalna ma zasadnicze znaczenie dla poprawy jakości życia pacjentów z zespołem Klinefeltera. W rzeczywistości, oprócz pokonywania deficytów fizycznych spowodowanych chorobą, poprawiają także ich integrację w życiu społecznym.
Rokowanie pogarsza się jednak dla tych, którzy nie są leczeni szybko lub są upośledzeni umysłowo. W szczególności u nieleczonych pacjentów, azoospermia, sterylność i małe jądra są nieodwracalnymi warunkami.


