ogólność
Kraniosynostoza jest terminem, w którym lekarze wskazują nieprawidłowość czaszki z powodu przedwczesnego zespolenia jednego lub więcej szwów czaszkowych.
Szwy czaszkowe to włókniste stawy łączące kości sklepienia czaszki (tj. Kości czołowe, skroniowe, ciemieniowe i potyliczne).
Kraniosynostoza może być izolowanym zjawiskiem (nie-syndromowa kraniosynostoza) lub wynikiem pewnych szczególnych stanów chorobowych (syndromiczna kraniosynostoza). Wśród chorób chorobowych, które powodują przedwczesną fuzję szwów czaszkowych, najbardziej znane są: zespół Crouzona i zespół Aperta.
Wraz z przedwczesnym stopieniem szwów czaszkowych, struktury mózgu nie mają odpowiedniej przestrzeni do wzrostu. Ma to różne konsekwencje, w tym głównie wzrost ciśnienia śródczaszkowego (nadciśnienie wewnątrzczaszkowe).
Szybka i dokładna diagnoza pozwala zaplanować leczenie ad hoc . Ten ostatni jest typu chirurgicznego i jego ostatecznym celem jest wczesne oddzielenie szwów bezpieczników.
Przypomina anatomię ludzkiej czaszki
Czaszka, wyposażona w kości i chrząstki, jest szkieletową strukturą głowy, która stanowi twarz i chroni mózg, móżdżek, pień mózgu i narządy zmysłów.

Aby uprościć badanie i zrozumienie czaszki, anatomowie pomyśleli o podzieleniu jej na dwa przedziały, zwane neurokranium i splancnocranium .
mózgoczaszka
Neurokranium to górny obszar czaszki, zawierający mózg i niektóre główne narządy zmysłów. Najważniejsze kości - ściśle płaskie - to kości czołowe, skroniowe, ciemieniowe i potyliczne; te razem wzięte tworzą tzw. sklepienie czaszki .
splanchnocranium
Splanchocranium, czyli masyw twarzy, jest przednio-dolnym obszarem czaszki, złożonym z równych i nierównych kości. Reprezentuje szkieletową strukturę twarzy, dlatego zawiera elementy kostne, takie jak żuchwa, górna szczęka, kości policzkowe, kość nosowa itp.
Czym jest kraniosynostoza?
Kraniosynostoza to rzadka anomalia czaszki, charakteryzująca się nienaturalnym kształtem głowy spowodowanym przedwczesnym stopieniem jednego lub więcej szwów czaszkowych . Szwy czaszkowe to włókniste stawy łączące kości sklepienia czaszki (tj. Kości czołowe, skroniowe, ciemieniowe i potyliczne).

Z witryny: //www.wkomsi.com/
KIEDY POWINIEN BYĆ ZAMKNIĘCIE KRUSZKOWYCH SZCZĘKÓW?
W normalnych warunkach fuzja szwów czaszkowych występuje w okresie poporodowym (uwaga: niektóre procesy kończą się nawet w wieku 20 lat). Ten długi proces fuzji umożliwia mózgowi prawidłowy wzrost i rozwój.
Jeśli, jak w przypadku kraniosynostozy, fuzja zachodzi zbyt wcześnie - dlatego w okresie prenatalnym, okołoporodowym * lub wczesnym dzieciństwie - elementy mózgu (mózg, móżdżek i pień mózgu) i niektóre narządy zmysłów (szczególnie oczy) ulegają zmiana kształtu i wzrostu.
przyczyny
Proces patofizjologiczny, który określa kraniosynostozę, to przedwczesne połączenie szwów czaszkowych .
Proces ten może stanowić izolowane zjawisko - gdzie przez „izolowany” rozumiemy, że nie jest on związany z żadnym konkretnym stanem chorobowym - lub może być konsekwencją pewnych szczególnych zespołów, prawie zawsze o charakterze genetycznym.
W związku z tym lekarze postanowili sklasyfikować kraniosynostozę na dwie kategorie:
- Non-syndromic craniosynostosis . Termin „nie-syndromowy” wskazuje, że anomalia czaszkowa nie jest związana z żadną patologią lub inną wadą fizyczną.
- Syndromiczna kraniosynostoza . Termin syndromiczny oznacza, że zniekształcenie czaszki jest wynikiem określonego zespołu, w większości przypadków typu genetycznego.
KRYZYSINOSTOSZA NIEZBĘDNA
Lekarze i badacze nie ustalili jeszcze, jakie są przyczyny nie-syndromowej kraniosynostozy.
Zaproponowali różne hipotezy - w tym wpływ czynników środowiskowych lub problemów typu hormonalnego - ale żadna z tych teorii nie znalazła potwierdzenia w wynikach eksperymentalnych.
Dlatego, aby zrozumieć dokładne pochodzenie anomalii, potrzebne są dalsze badania w tym zakresie.
CRANIOSINOSTOSI SINDROMICA
Zgodnie z najnowszymi badaniami medycznymi istnieje ponad 150 różnych zespołów, z których wszystkie są dość rzadkie i mogą powodować kraniosynostozę.
Wśród tych syndromów najbardziej znane i najpowszechniejsze są:
- Zespół Crouzona . Wynik specyficznych mutacji w genach FGFR2 (chromosom 10) i FGFR3 (chromosom 4), ten szczególny stan chorobowy dotyka jednego noworodka co 60 000 i prowadzi do występowania anomalii wyłącznie na poziomie głowy i twarzy.
- Zespół Aperta . Wynika to głównie z mutacji w genie FGFR2 (takich samych jak w zespole Crouzona) i dotyczy jednego noworodka co 100 000.
W przeciwieństwie do zespołu Crouzona, zmiany genetyczne FGFR2 są takie, że wady rozwojowe obejmują nie tylko czaszkę i twarz, ale także dłonie i stopy.
- Zespół Pfeiffera . Powstaje w wyniku mutacji „zwykłego” genu FGFR2 i genu o podobnych funkcjach, zwanego FGFR1 (chromosom 8). Osobliwością tych mutacji jest to, że - oprócz deformacji czaszki i twarzy - określają również: syndykty, brachydaktyty i kciuki oraz duże palce u nogi (nieproporcjonalne do pozostałych palców).
Zespół Pfeiffera występuje w jednym przypadku na 100 000 noworodków.
- Zespół Saethre'a-Chotzena . Jest to choroba genetyczna, która dotyka noworodka co około 50 000 osób. Powoduje różne deformacje czaszki, twarzy, rąk i stóp. Niektóre specyficzne mutacje genu TWIST1, znajdujące się na chromosomie 7, są odpowiedzialne za wystąpienie zespołu Saethre-Chotzen.
EPIDEMIOLOGIA CRANIOSINOSTOSI
Na podstawie najnowszych statystyk wydaje się, że dziecko cierpi na kraniosynostozę co około 1800-3000 lat.
Jeśli chodzi o najbardziej dotkniętą płeć, kilka badań klinicznych wykazało, że 3 na 4 pacjentów to mężczyźni. Powód, dla którego kraniosynostoza jest bardziej rozpowszechniona w populacji mężczyzn, jest całkowicie nieznany.
Czynniki ryzyka kraniosynostozy.
- Niska masa urodzeniowa
- Przedwczesne narodziny
- Zaawansowany wiek ojca
- Palenie przez matkę w czasie ciąży
Objawy i powikłania
Większość objawów obserwowanych w obecności kraniosynostozy wynika ze wzrostu ciśnienia wewnątrz czaszki . W medycynie wzrost ciśnienia wewnątrz czaszki nazywany jest nadciśnieniem śródczaszkowym lub nadciśnieniem śródczaszkowym .
W obecności kraniosynostozy nadciśnienie wewnątrzczaszkowe jest konsekwencją faktu, że mózg i inne struktury wewnątrz czaszki nie mają odpowiedniej przestrzeni do wzrostu, więc pchają struktury kostne głowy.
To powiedziawszy, ważne jest, aby pamiętać, że jeśli wiele szwów czaszkowych jest zaangażowanych lub jeśli stan nie jest leczony na czas, kraniosynostoza może prowadzić do zmniejszonego rozwoju zdolności poznawczych i niskiego IQ.
OBJAWY NADCIŚNIENIA ENDOCRANICZNEGO
Możliwe objawy nadciśnienia śródczaszkowego to:
- Trwały ból głowy. Na ogół pogarsza się rano i wieczorem.
- Problemy ze wzrokiem. Składają się z podwójnego widzenia, niewyraźnego widzenia i niewyraźnego widzenia.
- wymioty
- drażliwość
- Wypuchnięte lub wydatne oczy
- Trudności w śledzeniu ruchu przedmiotów
- Problemy ze słuchem
- Problemy z oddychaniem
- Zmiany stanu psychicznego
- papilledema
Liczba szwów czaszkowych zaangażowanych w rozwój kraniosynostozy ma decydujący wpływ na obecność nadciśnienia śródczaszkowego.
Na przykład lekarze zauważyli, że zaangażowanie pojedynczego szwu czaszkowego wywołuje nadciśnienie wewnątrzczaszkowe u 15% pacjentów; podczas gdy zaangażowanie co najmniej dwóch szwów prowadzi do wzrostu ciśnienia wewnątrz czaszki u co najmniej 60% pacjentów.
W obecności łagodnej postaci kraniosynostozy nadciśnienie wewnątrzczaszkowe zaczyna być problematyczne, powodując wspomnianą symptomatologię, około 4-8 lat życia.
ZNAKI CRANIOSINOSTOSI
Wśród objawów kraniosynostozy najczęściej występują:- Formacje sztywnych grzbietów wzdłuż szwów czaszkowych
- Nieprawidłowości czaszek czaszkowych
- Głowa o wymiarach nieproporcjonalnych do reszty ciała
RODZAJE KRANIOSINOSTOSIS
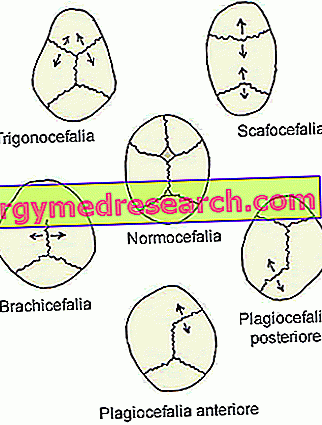
Kształt głowy pacjentów z kraniosynostozą zależy od tego, które szwy czaszkowe zamknęły się przedwcześnie.
Zauważywszy to, lekarze uznali, że należy odróżnić kraniosynostozę w różnych typach, w zależności od stosowanych szwów czaszkowych.
Rodzaje kraniosynostozy to:
- Synostoza strzałkowa ( dolichocephaly lub scaphocephaly ). Jest to najczęstszy rodzaj kraniosynostozy; w rzeczywistości charakteryzuje się około połową przypadków klinicznych.
Jego obecność zbiega się z przedwczesnym zamknięciem strzałkowych szwów czaszkowych, znajdujących się w górnej części czaszki, między kośćmi ciemieniowymi.
From //en.wikipedia.org/wiki/Plagiocephaly
- Kraniosynostoza koronalna ( brachycefalia ) Jest to drugi najczęściej występujący rodzaj kraniosynostozy; charakteryzuje około jednego przypadku klinicznego co cztery.
Jego początek obejmuje przedwczesną fuzję szwów koronalnych, które biegną między kością czołową a kośćmi ciemieniowymi.
- Metotopowa synostoza ( trigonocephaly ). Jest to bardzo rzadki typ kraniosynostozy, który wyróżnia tylko 4-10% przypadków.
Jego wygląd zbiega się z przedwczesnym stopieniem szwu metopowego (lub czołowego), który przebiega od nosa do górnej części głowy, oddzielając kość czołową na dwie części. Ogólnie rzecz biorąc, szew ten naturalnie ulega kostnieniu w szóstym roku życia.
- Synostoza lambdoidowa ( plagiocefalia ). Jest to najrzadszy rodzaj kraniosynostozy. W rzeczywistości jest to tylko 2-4% przypadków klinicznych.
Jego obecność polega na wczesnym zespoleniu szwu lambdoidowego, znajdującego się między kośćmi ciemieniowymi a kością potyliczną, z tyłu głowy.

POWIKŁANIA
Oprócz osłabienia rozwoju intelektualnego, nieleczona kraniosynostoza może określić:
- Tak zwany obturacyjny zespół bezdechu sennego .
- Trwałe zmiany twarzy, zwłaszcza w oczach i uszach.
- Stałe deformacje u podstawy czaszki (na przykład wady rozwojowe lub zespół Arnolda-Chiariego).
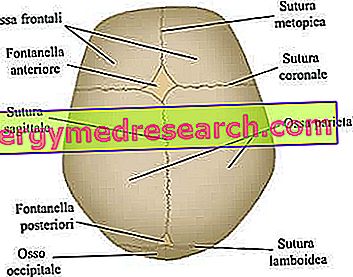
Główne szwy czaszkowe biorące udział w procesie kraniosynostozy. Z witryny: www.sciencebasedmedicine.org
- Wodogłowie .

diagnoza
Do zdiagnozowania kraniosynostozy, badania fizykalnego, oceny historii klinicznej i zdjęć radiologicznych dostarczonych przez promieniowanie rentgenowskie lub CT do głowy są niezbędne.
Jeśli kraniosynostoza jest typu syndromowego, ważne jest również ustalenie stanu chorobowego, który doprowadził do jej wystąpienia. Dlatego lekarze mogą skorzystać z badań krwi, a przede wszystkim z poradnictwa genetycznego .
CEL BADANIA
Badanie fizykalne polega na dokładnej analizie przez lekarza objawów klinicznych występujących na głowie pacjenta, podejrzanych o cierpienie na kraniosynostozę.
Ogólnie rzecz biorąc, pediatra jest odpowiedzialny za przeprowadzenie tej ważnej kontroli diagnostycznej.
HISTORIA KLINICZNA
Ocena historii klinicznej jest ważna dla celów diagnostycznych, ponieważ obejmuje pytania związane z czynnikami ryzyka kraniosynostozy.
Dlatego lekarz (zwykle zawsze pediatra) zbada, czy:
- Dziecko rodzi się przedwcześnie lub ma niską wagę.
- Jaki był wiek ojca w momencie poczęcia.
- Jeśli matka paliła podczas ciąży.
BADANIA RADIOLOGICZNE
Promieniowanie rentgenowskie i CT w głowie służą przede wszystkim potwierdzeniu diagnozy i pokazaniu lekarzowi, które szwy czaszkowe zrosły się przedwcześnie.
Wiedza o tym, które szwy czaszkowe są zaangażowane, pozwala zaplanować najbardziej odpowiednie leczenie chirurgiczne.
leczenie
Kraniosynostoza jest uleczalna tylko dzięki interwencji chirurgicznej .
Ta ostatnia składa się z operacji rozdzielania szwów czaszkowych łączących się wcześnie między nimi.
Ostatecznym celem terapeutycznym chirurgii jest dostarczenie struktur mózgowych i niektórych narządów zmysłów, takich jak oczy, z tą przestrzenią potrzebną do rozwoju i funkcjonowania w najlepszym wydaniu.
NAJLEPSZY CZAS NA INTERVENE
Nie ma całkowitej zgody lekarzy co do najlepszego czasu na operację kraniosynostozy.
Według niektórych ekspertów idealnym okresem dla operacji byłoby późne dzieciństwo, kiedy ryzyko nawrotu jest mniejsze (tj. Drugie przedwczesne zespolenie szwów czaszkowych). W przypadku nawrotu, operacja musi być powtórzona i nie jest to zalecane, biorąc pod uwagę delikatność procedury.
Według innych ekspertów najwłaściwszym czasem będzie wczesne dzieciństwo (między 6 a 12 miesiącem życia), kiedy czaszka nie jest jeszcze całkowicie skostniała, a kości nadal można formować. Możliwość kształtowania kości (plastyczność) pozwala na usunięcie wszelkich morfologicznych nieprawidłowości kości, co może spowodować poważne wady estetyczne i problemy funkcjonalne (do szczęki lub oczu) w bardziej dojrzałym wieku.
MOŻLIWE PODEJŚCIA CHIRURGICZNE
Istnieją dwa różne podejścia chirurgiczne: tradycyjna chirurgia, zwana również „otwartą”, oraz chirurgia endoskopowa.
- Tradycyjna (lub „otwarta”) operacja .
Przewiduje znieczulenie ogólne (dlatego pacjent jest nieprzytomny podczas całej operacji) oraz praktykę nacięcia chirurgicznego na głowie, dokładnie w punkcie, w którym obrazy radiologiczne wykazały anomalię czaszkową.
Poprzez nacięcie na głowie chirurg operacyjny (neurochirurg) usuwa nieprawidłową kość i powierza ją specjaliście w chirurgii twarzoczaszki, który ją modyfikuje i nadaje jej kształt umożliwiający normalny rozwój struktur mózgu.
Po modyfikacji neurochirurg ponownie umieszcza kość w jej pierwotnym położeniu i zamyka nacięcie szwami.
Podobnie jak wiele tradycyjnych operacji, operacja „otwarta” jest nieco inwazyjna; jednak, aby było to korzystne, fakt, że można modyfikować strukturę kości dokładnie iz dobrymi wynikami.
- Interwencja chirurgii endoskopowej .
Polega ona na użyciu endoskopu, narzędzia podobnego do elastycznej rurki, wyposażonego w kamerę światłowodową, na jednym końcu i podłączonego do monitora.
Z operacyjnego punktu widzenia polega to na wprowadzeniu endoskopu do otworu wykonanego na czaszce oraz w separacji, za pomocą samego endoskopu, szwów bezpieczników przedwcześnie.
Neurochirurgowi udaje się zorientować w głowie dzięki obrazom wyświetlanym przez kamerę na zewnętrznie podłączonym monitorze.
Interwencja chirurgii endoskopowej jest zdecydowanie mniej inwazyjna niż operacja „otwarta” (okres hospitalizacji jest również krótszy), jednak ma dwie wady: jest wskazany tylko dla pacjentów przez kilka miesięcy (ogólnie 6), które posiadają kształtne kości; jest większe ryzyko nawrotu.
FAZA PO-OPERACYJNA
Ogólnie rzecz biorąc, pacjent z kraniosynostozą, poddany zabiegowi chirurgicznemu, musi pozostać w szpitalu przez około 4-5 dni po operacji. W tym czasie neurochirurg i jego pracownicy okresowo monitorują swoje parametry życiowe i sprawdzają, czy wszystko idzie dobrze.
Po rezygnacji zaplanowano serię okresowych badań kontrolnych, które początkowo odbywają się co pół roku, a następnie wraz ze wzrostem pacjenta, co roku.
rokowanie
Rokowanie zależy od różnych czynników, w tym:
- Powoduje to wywołanie kraniosynostozy. Niektóre choroby genetyczne odpowiedzialne za tę anomalię są bardzo poważne i mają złe rokowanie.
- Pozycja szwów połączyła się wcześnie. Jeśli szwy znajdują się w pozycjach, do których neurochirurg jest „niewygodny” do osiągnięcia, interwencja kraniosynostozy staje się skomplikowana i może nie zapewniać pożądanych rezultatów.


