ogólność
Zespół Pradera-Williego jest rzadkim zaburzeniem genetycznym, które powoduje zaburzenia fizyczne, behawioralne i intelektualne. Najbardziej charakterystycznymi objawami klinicznymi są otyłość (i związane z nią choroby) oraz zmniejszone napięcie mięśniowe.
Badanie fizykalne jest zwykle wystarczające do ustalenia prawidłowej diagnozy, ale można również przeprowadzić wiarygodne testy genetyczne.
Niestety nadal nie ma definitywnej terapii; jednak niektóre farmakologiczne i behawioralne środki zaradcze mogą ograniczać powiązaną symptomatologię.
Krótkie przypomnienie genetyki
Przed opisaniem zespołu Pradera-Williego warto krótko odnieść się do genetyki.
CHROMOSOMY I DNA
Każda komórka zdrowego człowieka ma 23 pary homologicznych chromosomów : 23 są matczyne, czyli odziedziczone po matce, a 23 są ojcowskie lub odziedziczone po ojcu. Para tych chromosomów ma charakter seksualny, to znaczy określa płeć jednostki; pozostałe 22 pary składają się z chromosomów autosomalnych . W sumie 46 ludzkich chromosomów zawiera cały materiał genetyczny, lepiej znany jako DNA . W DNA osoby zapisywane są jego cechy somatyczne, predyspozycje, zdolności fizyczne itp.
GENY I MUTACJE DNA
DNA jest zorganizowany w wiele sekwencji, mniej lub bardziej długich, zwanych genami .

Rysunek: organizacja genu w parze homologicznych chromosomów. Para homologicznych chromosomów zawiera specyficzne geny, wszystkie mające dwa warianty, allele, zajmujące tę samą pozycję chromosomalną i wykonujące te same funkcje (z wyjątkiem mutacji). Lewa para chromosomów ma dwa równe allele (oba niebieskie); prawa para ma dwa różne allele (jeden jest czerwony, a drugi niebieski).
Każdy gen zajmuje określony chromosom i jego odpowiednik, ponieważ występuje w dwóch kopiach, zwanych allelami . Allel pochodzi od matki i znajduje się w chromosomie matki; drugi allel pochodzi od ojca i jest trzymany w chromosomie ojcowskim.
Z genów, z których pochodzą białka, obecnych w naszym ciele. Gdy zachodzi mutacja DNA, gen (zwykle allel) danego chromosomu może być wadliwy, a zatem może wytwarzać wadliwe białko lub całkowicie go brakować.
Co to jest zespół Pradera-Williego?
Zespół Pradera-Williego jest rzadkim zaburzeniem genetycznym charakteryzującym się szeregiem deficytów fizycznych, intelektualnych i behawioralnych zależnych od zmiany chromosomu 15.
We wczesnym dzieciństwie zespół manifestuje się niezwykłym osłabieniem mięśni i opóźnieniem rozwoju. Później, w dzieciństwie, pojawiają się inne problemy, takie jak ciągły apetyt, trudności w nauce i anomalie behawioralne.
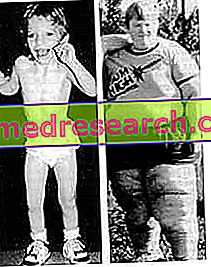
Osoby z zespołem Pradera-Williego są bardzo często osobami cierpiącymi na otyłość, z których wynikają różne problemy sercowe. Są to główna przyczyna śmierci.
epidemiologia
Zespół Pradera-Williego jest rzadką chorobą: w rzeczywistości narodziny chorego dziecka rejestruje się średnio co 15 000–30 000 noworodków.
Wpływa na mężczyzn i kobiety w równym stopniu i nie ma predylekcji dla niektórych ras.
przyczyny
Przyczyną powodującą zespół Pradera-Williego jest mutacja genetyczna na chromosomie 15 . Dokładny gen uderzony nie został jeszcze wyjaśniony; podejrzenia spadają bardziej niż cokolwiek innego na region chromosomalny, który obejmuje więcej genów.

Rysunek: chromosom 15 i geny podejrzewane o rolę w zespole Pradera-Williego. Z witryny: www.kreatech.com
GENY NA CHROMOSOMIE 15
Zwykle komórki naszego ciała wykorzystują oba allele do tworzenia białek. Innymi słowy, oznacza to, że oba chromosomy, matczyne i ojcowskie, są użyteczne i zapewniają swój wkład genetyczny.
Jednak w niektórych konkretnych komórkach, z powodu ewolucyjnego i niepatologicznego problemu, działa tylko jeden allel ojcowski lub matczyny, a jego praca jest więcej niż zadowalająca. Nieaktywny allel nazywany jest cichym, właśnie dlatego, że istnieje, ale sam się nie „wyraża”.
Wszystkie komórki naszego mózgu zawierają chromosom 15, ale w niektórych regionach wyrażana jest tylko matczyna linia genetyczna, podczas gdy w innych tylko ojcowska. W podwzgórzu, który jest obszarem mózgu odpowiedzialnym za zespół Pradera-Williego, tylko geny chromosomu ojcowskiego są normalnie wyrażane.
CHROMOSOMA 15 I SYNDROM PRADER-WILLI
Dzięki testom genetycznym stwierdzono, że pacjentom z zespołem Pradera-Williego brakuje normalnego chromosomu ojcowskiego 15. Jest to szkodliwe w podwzgórzu, gdzie jedyną aktywną linią chromosomalną jest linia ojcowska.
Główne funkcje podwzgórza:
- Regulacja apetytu
- Dostosowywanie rytmów uśpienia i czuwania
- Ekspresja stanów emocjonalnych
- Regulacja temperatury ciała
- Produkcja hormonów
Ale co powoduje nieprawidłowe funkcjonowanie lub brak chromosomu ojcowskiego 15? Istnieją co najmniej trzy możliwe przyczyny:
- Brak konkretnego regionu chromosomu ojcowskiego 15: w rzeczywistości chromosomowi brakuje istotnej części.
- Dwa chromosomy 15 matczyne. Ta anomalia występuje z powodu błędu podczas tworzenia zarodka.
- Zmiana niektórych genów obecnych na chromosomie ojcowskim 15.
GENETYCZNE I HEREDITARY? LUB TYLKO GENETYKA?
Genetycy uważają zespół Pradera-Williego za chorobę genetyczną, ponieważ według badań okazało się, że trzy wyżej wymienione tryby mutacji nie są dziedziczone od rodziców (którzy posiadają normalny strój chromosomalny), ale powstają przypadkowo, tuż przed poczęciem ( sporadyczna mutacja ).
Jednak prawdziwość tego stwierdzenia jest podważana przez wykrycie niektórych par rodziców z więcej niż jednym dzieckiem cierpiącym na zespół Pradera-Williego. W tych przypadkach istnieje powód, by sądzić, że u źródła choroby może być jakiś element dziedziczny, który należy jeszcze wykazać.
SYNDROM PRADER-WILLI I ZESPÓŁ ANGELMANA
Zespół Pradera-Williego jest w pewnym sensie przeciwieństwem zespołu Angelmana : w tym drugim przypadku chromosom matczyny 15 nie działa prawidłowo.
Objawy i powikłania
Aby dowiedzieć się więcej: Objawy zespołu Pradera-Williego
Zespół Pradera-Williego objawia się, wraz z pierwszymi objawami i objawami, już w najwcześniejszym niemowlęctwie (pierwszy rok życia); w tym okresie powoduje głównie zmniejszenie napięcia mięśniowego (hipotonia) i opóźnienie rozwoju. Z biegiem czasu choroba przechodzi pewnego rodzaju ewolucję, która dodatkowo wzbogaca symptomatyczny obraz.
DZIECIŃSTWO
Główne znaki w pierwszym roku życia to:
- Mięśniowa hipotonia . Oznacza to, że ton mięśni jest zmniejszony w porównaniu do normalnego: zwykle objawia się miękkimi i mało reaktywnymi kończynami, a także trudnym ssaniem mleka matki.
- Opóźnienie rozwoju . Sprzyja temu ssanie z powodu hipotonii.
- Zez .
- Charakterystyczne rysy twarzy . Oczy migdałowe, zwężenie głowy w skroniach, usta w dół i cienka górna warga.
- Częściowy lub całkowity brak odpowiedzi na bodźce . Dziecko jest zmęczone i nie jest łatwo go obudzić.
OD WIEKU DO WIEKU? DLA DOROSŁYCH
Od pierwszego roku życia pojawiła się długa seria problemów, które mogą mieć dramatyczne skutki.
- Niezwykły apetyt i otyłość . Pacjenci wykazują ciągłe pragnienie jedzenia, co powoduje, że dużo jedzą i zyskują znaczną wagę. Jeśli nie znajdą nic do jedzenia, przychodzą, by spożywać zamrożone jedzenie i odpady, innymi słowy, wszystko, co jadalne. Wszystko to wynika ze zmienionych funkcji podwzgórza.
- Hipogonadyzm . Oznacza to, że narządy płciowe (jądra, u ludzi i jajniki u kobiet) wytwarzają niewiele hormonów płciowych (męski testosteron i żeński estrogen). Pacjent nie kończy rozwoju dojrzewania i zwykle nie jest płodny. Pierwsza miesiączka u kobiet jest opóźniona (jeśli nie całkowicie nieobecna); u ludzi nie obserwuje się zmiany głosu.
- Zmniejszony wzrost i rozwój . Do problemu hipotonii mięśniowej, która pozostaje, dodaje się zmniejszony rozwój staturalny, nawet po okresie dojrzewania (w którym zazwyczaj dorastają kilka centymetrów).
- Deficyty uczenia się . Intelektualne zdolności pacjentów są prawie zawsze zmniejszone.
- Problemy behawioralne . Zwłaszcza w okresie dojrzewania jednostki są uparte, kapryśne i cierpią na tak zwane zaburzenie obsesyjno-kompulsyjne.
- Opóźnienie silnika . Dzieci uczą się chodzić bardzo późno.
- Trudności językowe . Zazwyczaj pacjenci zaczynają rozmawiać ze znacznym opóźnieniem, a ich język zawsze pozostaje słaby i trudny.
- Zaburzenia snu . Normalna przemiana faz snu REM i NON-REM nie jest przestrzegana. Ponadto pacjenci, gdy śpią, cierpią z powodu przerw w oddychaniu (bezdech senny).
- Skolioza . Problem jest zarezerwowany tylko dla niektórych pacjentów.
KIEDY ZOBACZYĆ SIĘ DO LEKARZA
W niemowlęcia. Objawy, które powinny skłonić do podejrzenia zespołu Pradera-Williego to: brak rozwoju, hipotonia mięśniowa, trudności w ssaniu mleka, rysy twarzy i brak reakcji na bodźce.
W dziecku. Dwie podstawowe wskazówki to: uporczywe poszukiwanie pożywienia i szybki wzrost wagi.
POWIKŁANIA
Główne powikłania zespołu Pradera-Williego wynikają z otyłości i wszystkich związanych z nią problemów, takich jak cukrzyca, choroby serca, nadciśnienie, hipercholesterolemia, miażdżyca itp. Ponadto, pozostając w kontekście ciągłego odżywiania, pacjentowi łatwo jest udusić się z powodu posiłku spożywanego z żarłocznością.
Kolejna seria bardzo ważnych powikłań jest związana z hipogonadyzmem : brak hormonów płciowych bardzo często powoduje bezpłodność i osteoporozę .
diagnoza
Przed skorzystaniem z testów genetycznych, prawidłowe wstępne rozpoznanie zespołu Pradera-Williego można również ustalić za pomocą prostego badania fizykalnego i niektórych badań krwi.
Objawy kliniczne, które należy znaleźć podczas badania fizykalnego
u niemowlęcia:
- Mięśniowa hipotonia
- Migdałowe oczy
- Kurczenie się skroni
W dziecku / nastolatku:
- Nienasycony apetyt
- otyłość
- Problemy behawioralne
Testy genetyczne służą jako potwierdzenie i pomagają wyjaśnić rodzaj mutacji, która spowodowała chorobę.
leczenie
Niestety, ponieważ jest to choroba genetyczna, zespół Pradera-Williego nie jest uleczalny.
Jedyne stosowane zabiegi terapeutyczne to ograniczenie objawów (na przykład otyłości), złagodzenie niektórych nieprawidłowych zachowań i, ogólnie, poprawa standardu życia pacjentów.
Aby to wszystko osiągnąć, wskazane jest zwrócenie się do zespołu lekarzy i ekspertów, specjalizujących się w różnych dziedzinach, od endokrynologii po dietologię, od fizjoterapii po psychoterapię.
Najczęstsze środki terapeutyczne są wymienione poniżej.
ŻYWIENIE W DZIEDZINIE DZIECKA I W KOLEJNYCH KROKACH
We wczesnym dzieciństwie, aby przezwyciężyć trudności związane z ssaniem i brakiem rozwoju, dobrze jest dać dziecku posiłki wysokokaloryczne.
W następujących fazach sytuacja całkowicie się zmienia: podawane posiłki muszą być dokładnie sprawdzone, zwracając szczególną uwagę na kalorie.
Najlepszym specjalistą, który poprosi o radę, jest dietetyk .
HORMON WZROSTU
Egzogenne podawanie (tj. Z zewnątrz) hormonu wzrostu ( GH ) ma trzy efekty:
- Zachęcanie do wzrostu, którego inaczej by nie było
- Popraw napięcie mięśniowe
- Zmniejsz poziom tkanki tłuszczowej
Leczenie rozpoczyna się w wieku około 3-5 lat.
Obecnie w laboratorium wytwarzane są preparaty hormonalne, skuteczne i o ograniczonych skutkach ubocznych.
Najbardziej wskazanym specjalistą w tym przypadku jest endokrynolog .
HORMONY SEKSUALNE
Egzogenne podawanie testosteronu dla mężczyzn i estrogenów dla kobiet ma zasadnicze znaczenie dla przywrócenia obniżonego poziomu tych dwóch hormonów. Oprócz poprawy płodności terapia hormonalna ma również wpływ na osteoporozę.
Leczenie rozpoczyna się w okresie dojrzewania.
Aby dowiedzieć się więcej: Leki stosowane w leczeniu zespołu Pradera-Williego »
FIZJOTERAPIA I LOGOPEDIA
Pacjenci z zespołem Pradera-Williego potrzebują rehabilitacji fizycznej i językowej . Pierwszy ma na celu ograniczenie hipotonii mięśniowej i skutków otyłości; drugie lekarstwo na braki komunikacyjne, zarówno mówione, jak i pisane.
Eksperci, aby się zwrócić, są odpowiednio fizjoterapeutą i logopedą.
PSYCHOTERAPIA I TERAPIA ZATRUDNIENIA
Psychoterapia jest niezbędna dla pacjentów z zaburzeniami obsesyjno-kompulsyjnymi i ogólnie z nastrojem. Wsparcie psychiatry lub psychologa może znacznie poprawić aspekt behawioralny.
Z drugiej strony, terapia zajęciowa ma na celu nauczenie pacjenta, jak dbać o siebie, jak się ubierać itd., Innymi słowy, jak wykonywać główne codzienne czynności.
POMOC RODZIN
Bliskość członków rodziny jest niezbędna, aby pomóc chorym krewnym, zwłaszcza w młodości. Radzenie, które jest zazwyczaj udzielane rodzinom, to podążanie za pacjentem we wszystkich jego działaniach (zwłaszcza gdy jest nakarmiony), wypytywanie o najodpowiedniejsze zachowanie dla niego, aby nie wykluczać go itp.
Rokowanie i zapobieganie
Biorąc pod uwagę, że zespół Pradera-Williego jest chorobą nieuleczalną, rokowanie nigdy nie będzie pozytywne. Największym zagrożeniem jest otyłość i związane z nią patologie: śmierć jest zwykle spowodowana jednym z nich.
Dostępne zabiegi (zrównoważona dieta, terapia hormonalna, psychoterapia itp.) Poprawiają jakość życia, nawet w delikatny sposób; jednak nadal istnieją środki kontroli objawów i nic więcej.
Bliskość krewnych jest fundamentalna: ich wsparcie może w rzeczywistości przedłużyć życie pacjentów.
PROFILAKTYKA
Gdy choroba powstaje w embrionie z powodu mutacji genetycznej, nie ma sposobu, aby temu zapobiec.
Jeśli zamiast tego dwoje rodziców urodziło już dziecko z zespołem Pradera-Williego, przed drugą ciążą mogą przejść specjalne testy genetyczne, aby dowiedzieć się, czy są nosicielami choroby, czy nie.